Edited by Chi Hoang, MS; Tuan Tran, MD, PhD.
1. MÔ TẢ
Hội chứng Angelman là một rối loạn gen phức tạp, chủ yếu ảnh hưởng đến hệ thần kinh. Hội chứng Angelman ảnh hưởng đến khoảng 1 trong 12.000 đến 20.000 người. Các đặc điểm đặc trưng của tình trạng này bao gồm chậm phát triển, giảm thiểu trí tuệ (intellectual disability), suy giảm khả năng nói nghiêm trọngvà các vấn đề về vận động và thăng bằng (ataxia). Hầu hết trẻ em bị ảnh hưởng cũng bị co giật thường xuyên (epilepsy) và kích
thước đầu nhỏ (microcephaly). Sự phát triển bị trì hoãn trở nên đáng chú ý khi được 6 đến 12 tháng tuổi, và các dấu hiệu và triệu chứng phổ biến khác thường xuất hiện ở thời thơ ấu.

Trẻ em mắc hội chứng Angelman thường có thái độ vui vẻ, dễ bị kích động với những động tác cười và vỗ tay thường xuyên. Tăng động, một khoảng chú ý ngắn khả năng tập trung kém và niềm đam mê với nước là những dấu hiệu phổ biến. Hầu hết trẻ em bị ảnh hưởng cũng khó ngủ và cần ngủ ít hơn bình thường.
Với tuổi tác, những người mắc hội chứng Angelman trở nên ít hưng phấn phản ứng thái quá hơn và các vấn đề về giấc ngủ có xu hướng cải thiện. Tuy nhiên, những người bị ảnh hưởng tiếp tục bị thiểu năng trí tuệ, suy giảm khả năng nói nghiêm trọng và co giật trong suốt cuộc đời của họ. Người lớn mắc hội chứng Angelman có các đặc điểm trên khuôn mặt có thể được mô tả là “thô” (coarse). Các đặc điểm phổ biến khác bao gồm làn da trắng
bất thường với mái tóc sáng màu và một độ cong bất thường từ cột sống hoặc vẹo cột sống (scoliosis). Tuổi thọ của những người mắc bệnh này dường như gần như bình thường.
2. Nguyên nhân
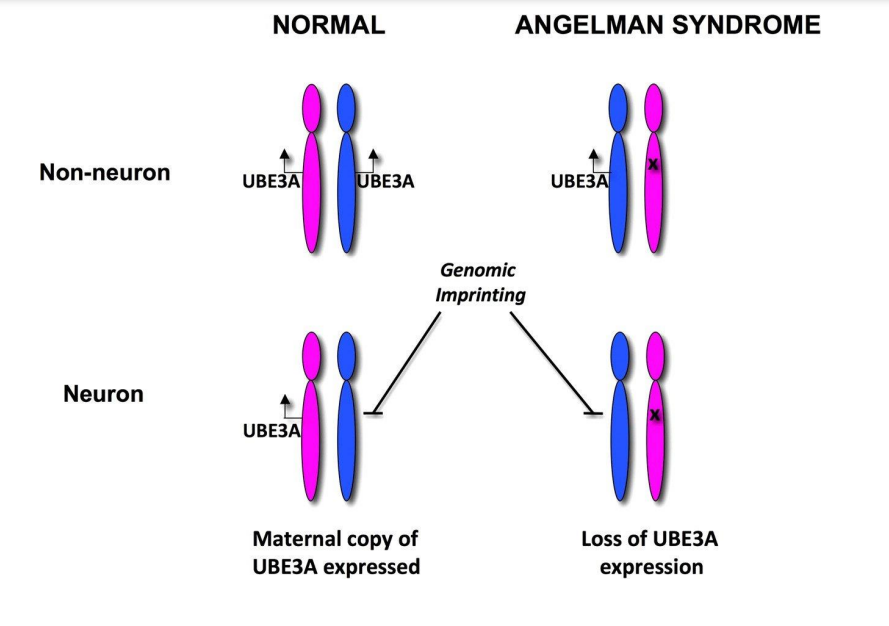
Nhiều đặc điểm đặc trưng của hội chứng Angelman là do mất chức năng của một gen tên là UBE3A. Mọi người thường thừa hưởng một bản sao của gen UBE3A từ mỗi cha mẹ. Cả hai bản sao của gen này được bật (hoạt động) trong nhiều mô của cơ thể. Tuy nhiên, trong một số khu vực nhất định của não, chỉ có bản sao được thừa hưởng từ mẹ (bản sao của mẹ) là hoạt động. Kích hoạt gen dành riêng cho cha mẹ này được gây ra bởi một hiện tượng gọi là dấu ấn gen (gene or genomic imprinting); cho nên nếu bản sao gen của mẹ trong một người bị mất do thay đổi nhiễm sắc thể hoặc đột biến gen, người đó sẽ không có bản sao gen hoạt động ở một phần của não.
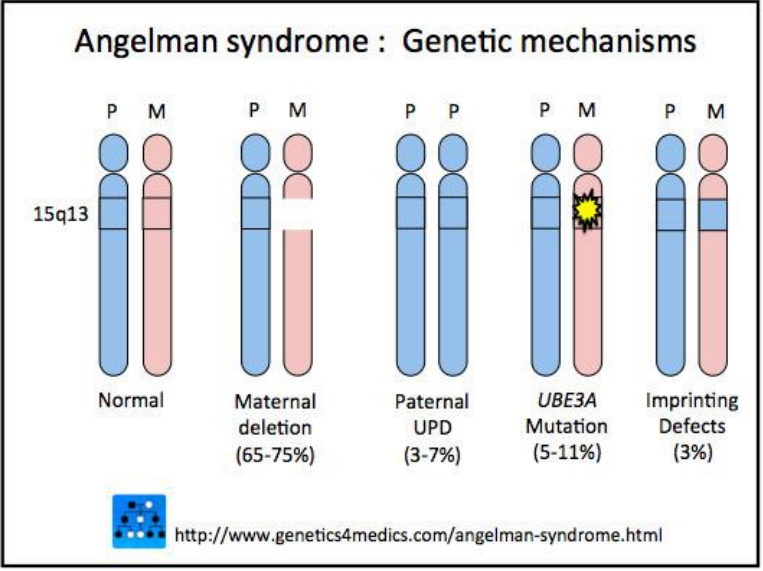
Hầu hết các trường hợp, hội chứng Angelman (khoảng 70%) xảy ra khi là do mất một đoạn nhiễm sắc thể 15 của mẹ có chứa gen này bị xóa. Trong các trường hợp khác (khoảng 10-15%), hội chứng Angelman được gây ra bởi một đột biến trong bản sao gen UBE3A của mẹ. Một tỷ lệ nhỏ hội chứng Angelman có khi một người được gây ra do người bệnh thừa hưởng hai bản sao nhiễm sắc thể 15 từ cha của họ (bản sao của cha) thay vì một bản sao từ mỗi cha mẹ. Hiện tượng này được gọi là đơn phụ song thể từ cha (paternal uniparental disomy). Hiếm khi, hội chứng Angelman cũng có thể được gây ra bởi sự sắp xếp lại nhiễm sắc thể được hay còn gọi là chuyển vị (translocation), hoặc do đột biến hoặc khiếm khuyết trong DNA kiểm soát kích hoạt gen UBE3A. Những thay đổi di truyền này có thể tắt (bất hoạt) UBE3A hoặc các gen khác của nhiễm sắc thể 15 của mẹ. Cuối cùng, 10-15% hội chứng Angelman chưa được biết nguyên nhân, những thay đổi liên quan đến các gen hoặc nhiễm sắc thể khác có thể là nguyên nhân gây ra rối loạn trong những trường hợp này.
Ở một số người mắc hội chứng Angelman, việc mất một gen tên OCA2 có liên quan đến mái tóc sáng màu và làn da trắng.
Gen OCA2 (trước đây gọi là gen P) hướng dẫn tạo ra một protein gọi là protein P. Protein này nằm trong melanocytes, là những tế bào chuyên biệt tạo ra sắc tố melanin. Melanin là chất mang lại màu da, tóc và mắt. Melanin cũng được tìm thấy trong các mô nhạy cảm với ánh sáng ở phía sau mắt (võng mạc), nơi nó đóng vai trò trong thị giác.
Mặc dù chức năng chính xác của protein P vẫn chưa được biết, nhưng nó rất cần thiết cho việc tạo ra sắc tố bình thường và có khả năng liên quan đến việc sản xuất melanin. Trong melanocytes, protein P có thể vận chuyển các phân tử vào và ra khỏi các cấu trúc gọi là melanosome (nơi sản xuất melanin).Protein này cũng có thể giúp điều chỉnh độ pH của melanosome.
Gen OCA2 nằm trên đoạn nhiễm sắc thể 15 thường bị xóa ởnhững người mắc hội chứng Angelman; tuy nhiên, mất gen OCA2 không gây ra các dấu hiệu và triệu chứng khác của hội chứng. Protein được tạo ra từ gen này giúp xác định màu sắc (sắc tố) của da, tóc và mắt.
Các nhà khoa học đang nghiên cứu các gen khác trên nhiễm sắc thể 15 cũng có thể liên quan đến các dấu hiệu và triệu chứngchính của tình trạng này.
3. Di truyền
Hầu hết các trường hợp mắc hội chứng Angelman không được di truyền, đặc biệt là những trường hợp gây ra bởi việc xóa nhiễm sắc thể 15 của mẹ hoặc do sự đơn phụ song thể từ cha (paternal uniparental disomy). Những thay đổi gen này xảy ra như sự kiện ngẫu nhiên trong quá trình hình thành các tế bào sinh sản (trứng và tinh trùng) hoặc trong sự phát triển phôi thai sớm. Những người bị ảnh hưởng thường không có tiền sử rối loạn trong gia
đình họ.
Hiếm khi, một thay đổi di truyền tạo ra hội chứng Angelman. Ví dụ, có thể có đột biến gen UBE3A hoặc trong khu vực DNA gần đó kiểm soát kích hoạt gen được truyền từ thế hệ này sang thế hệ tiếp theo.
4. XÉT NGHIỆM
• Fluorescence In Situ Hybridization (FISH-interphase, FISHmetaphase)
• Targeted variant analysis (Variant analysis in TruSightSoftware Suite begins with automatic alignment and variant calling using the DRAGEN Platform, followed by triaging,
visualizing, and interpreting variants)
• Deletion/duplication analysis: Disorder Gene Protein Inher Mod

Sequence analysis of the entire coding region
• Uniparental disomy study (UPD)
• Detection of homozygosity
• Methylation analysis
5. SOURCES
▪ Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010 Aug 15;154C(3):365-76. doi: 10.1002/ajmg.c.30273. Review.
▪ Dagli AI, Mueller J, Williams CA. Angelman Syndrome. 1998 Sep 15 [updated 2015 May 14]. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. Available from http://www.ncbi.nlm.nih.gov/books/NBK1144/
▪ Gentile JK, Tan WH, Horowitz LT, Bacino CA, Skinner SA, Barbieri-Welge R, Bauer-Carlin A, Beaudet AL, Bichell TJ, Lee HS, Sahoo T, Waisbren SE, Bird LM, Peters SU. A neurodevelopmental survey of Angelman syndrome with genotype-phenotype correlations. J Dev Behav Pediatr. 2010 Sep;31(7):592-601. doi: 10.1097/DBP.0b013e3181ee408e.
Erratum in: J Dev Behav Pediatr. 2011 Apr;32(3):267.
▪ Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cell Mol Life Sci. 2007 Apr;64(7-8):947-60. Review.
▪ Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, Hutson A, Nicholls RD, Zori RT, Williams CA, Driscoll DJ. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001 Dec;38(12):834-45.
▪ Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat. 2008 Jun;4(3):577-84.
▪ Tan WH, Bacino CA, Skinner SA, Anselm I, Barbieri-Welge R, Bauer-Carlin A, Beaudet AL, Bichell TJ, Gentile JK, Glaze DG, Horowitz LT, Kothare SV, Lee HS, Nespeca MP, Peters SU, Sahoo T, Sarco D, Waisbren SE, Bird LM. Angelman syndrome: Mutations influence features in early childhood. Am J Med Genet A. 2011 Jan;155A(1):81-90. doi:
10.1002/ajmg.a.33775.
▪ Van Buggenhout G, Fryns JP. Angelman syndrome (AS, MIM 105830). Eur J Hum Genet. 2009 Nov;17(11):1367-73. doi:10.1038/ejhg.2009.67. Epub 2009 May 20. Review.
▪ Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006 Mar 1;140(5):413-8.
▪ Rett/Angelman Syndromes and Related Disorders Panel Sequence Analysis and Exon-Level Deletion/Duplication Testing Of 12 Genes. Panel Gene List: CDKL5, CNTNAP2,
FOXG1*, MBD5, MECP2, MEF2C, NRXN1, SLC9A6, TCF4, UBE3A, WDR45, ZEB2 *This panel does not include deletion/duplication testing of FOXG1
▪ Williams CA. Neurological aspects of the Angelman syndrome. Brain Dev. 2005 Mar;27(2):88-94. Review.
▪ Williams CA. The behavioral phenotype of the Angelman
syndrome. Am J Med Genet C Semin Med Genet. 2010 Nov 15;154C(4):432-7. doi: 10.1002/ajmg.c.30278. Review.
▪ Gillfillan et al., (2008) Am J Hum Genet 82(4):1003-1010
▪ de Pontual et al., (2009) Hum Mutat 30:669-676.
▪ Hodge et al., (2013) Mol Psych Apr 16. doi: 10.1038/mp.2013.42.
Trân trọng cảm ơn!
{kind=link}